
Journal of South China University of Technology(Natural Science) >
Prediction of Target Inhibitor Activity by Integrating Machine Learning and Metaheuristic Algorithms
Received date: 2025-01-17
Online published: 2025-11-27
Supported by
the National Natural Science Foundation of China(12322119)
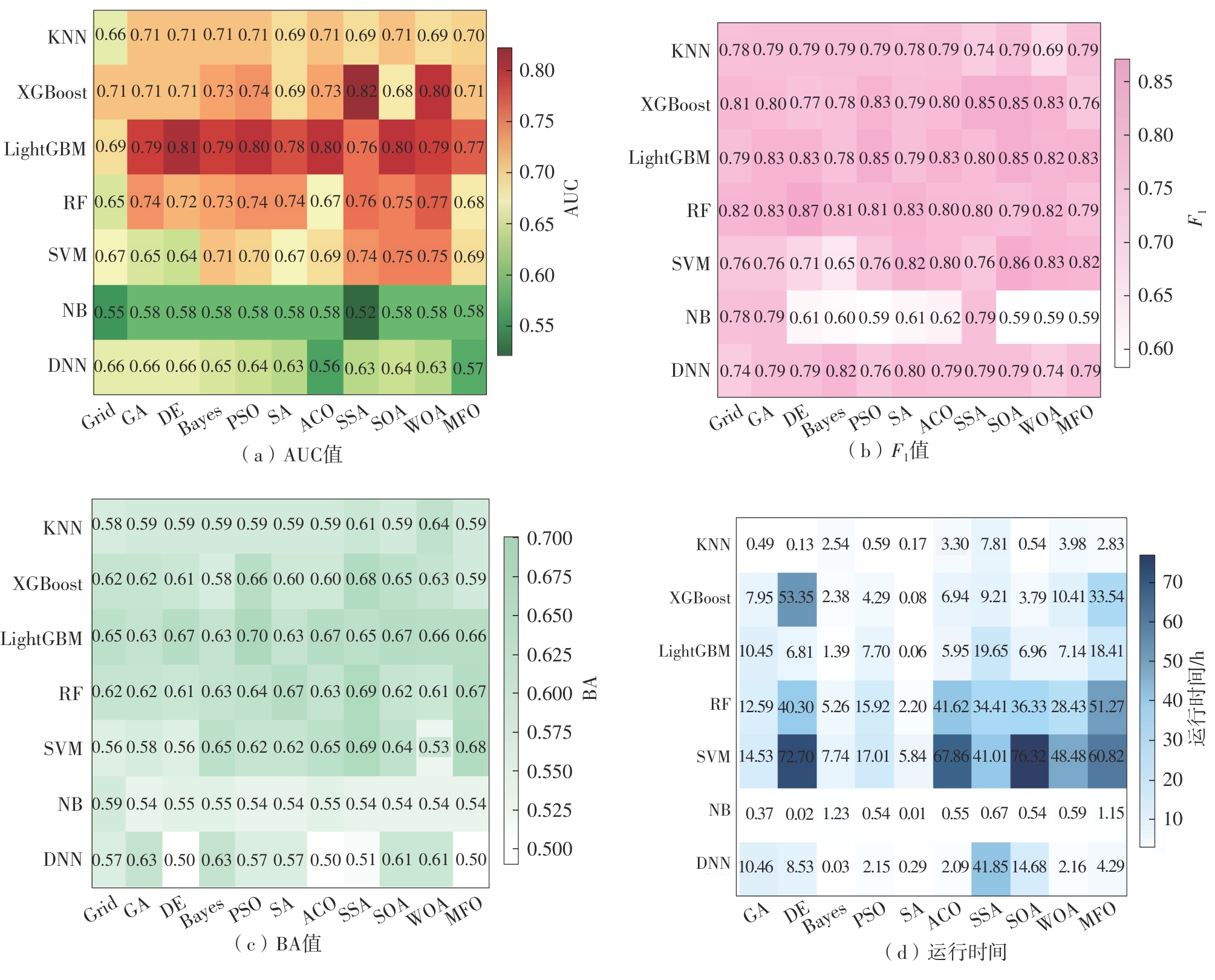
Traditional machine learning (ML) and deep learning (DL) play a key role in predicting the activity of target inhibitors. Many models based on existing datasets can predict compound bioactivity. However, debate persists regarding whether ML or DL performs better for such prediction tasks. In this study, datasets were constructed based on different molecular representations. Ten metaheuristic algorithms were applied to optimize the hyperparameters of eleven ML and DL models, aiming to systematically compare their predictive performance and identify the optimal ones. The results show that ML and DL models whose hyperparameters were optimized by metaheuristic algorithms significantly outperformed those optimized using the traditional grid search method. Furthermore, in low-dimensional feature spaces, graph-based DL models, such as SSA-GAT and SSA-Attentive FP, can automatically extract informative features from data via an end-to-end learning mechanism, yielding better performance than ML models. In contrast, in high-dimensional feature spaces (e.g., the feature space formed by combining RDKit descriptors with ECFP, AtomPairs, and MACCS fingerprints), ML methods, leveraging the complementary information in molecular features and the powerful optimization capability of metaheuristic algorithms, can effectively capture complex feature interactions. Consequently, ML methods often demonstrate higher accuracy and robustness in high-dimensional modeling. These findings provide valuable guidance for selecting between ML and DL approaches for target inhibitor activity prediction.
LING Fei , GU Xuerong . Prediction of Target Inhibitor Activity by Integrating Machine Learning and Metaheuristic Algorithms[J]. Journal of South China University of Technology(Natural Science), 2026 , 54(2) : 91 -101 . DOI: 10.12141/j.issn.1000-565X.250020
| [1] | FENG B, LIU Z, HUANG N,et al .A bioactivity foundation model using pairwise meta-learning[J].Nature Machine Intelligence,2024,6(8):962-974. |
| [2] | ZHANG H, MAO J, QI H Z,et al .Developing novel computational prediction models for assessing chemical-induced neurotoxicity using na?ve Bayes classifier technique[J].Food and Chemical Toxicology,2020,143:111513/1-11 |
| [3] | PRABHA N K, SHARMA A, SANDHU H,et al .TNFipred:a classification model to predict TNF-α inhibitors[J].Molecular Diversity,2024,28(3):1697-1707. |
| [4] | LI B, KANG X, ZHAO D,et al .Machine learning models combined with virtual screening and molecular docking to predict human topoisomerase I inhibitors[J].Molecules,2019,24(11):2107/1-16. |
| [5] | SHI J, ZHAO G, WEI Y .Computational QSAR model combined molecular descriptors and fingerprints to predict HDAC1 inhibitors[J].Medecine Sciences,2018,34:52-58. |
| [6] | KANG M G, KANG N S .Predictive model for drug-induced liver injury using deep neural networks based on substructure space[J].Molecules,2021,26(24):7548/1-16 |
| [7] | DENG J, YANG Z, WANG H,et al .A systematic study of key elements underlying molecular property prediction[J].Nature Communications,2023,14(1):6395/1-20. |
| [8] | LV Q, CHEN G, YANG Z,et al .Meta learning with graph attention networks for low-data drug discovery[J].IEEE Transactions on Neural Networks and Learning Systems,2023,35(8):11218-11230. |
| [9] | YANG K, SWANSON K, JIN W,et al .Analyzing learned molecular representations for property prediction[J].Journal of Chemical Information and Modeling,2019,59(8):3370-3388. |
| [10] | MASTROPIETRO A, PASCULLI G, BAJORETH J .Learning characteristics of graph neural networks predicting protein-ligand affinities[J].Nature Machine Intelligence,2023,5(12):1427-1436. |
| [11] | MAYR A, KLAMBAUER G, UNTERTHINER T,et al .Large-scale comparison of machine learning methods for drug target prediction on ChEMBL[J].Chemical Science,2018,9:5441-5451. |
| [12] | WAINER J, FONSECA P .How to tune the RBF SVM hyperparameters?An empirical evaluation of 18 search algorithms[J].Artificial Intelligence Review,2021,54(6):4771-4797. |
| [13] | BERGSTRA J, BENGIO Y .Random search for hyper-parameter optimization[J].Journal of Machine Lear-ning Research,2012,13(2):281-305. |
| [14] | WANG X, JIN Y, SCHMITT S,et al .Recent advances in Bayesian optimization[J].ACM Computing Surveys,2023,55(13s):1-36. |
| [15] | YAMASHITA R, NISHIO M, DO R K G,et al .Convolutional neural networks:an overview and application in radiology[J].Insights into Imaging,2018,9:611-629. |
| [16] | CERETO-MASSAGUé A, OJEDA M J, VALLS C,et al .Molecular fingerprint similarity search in virtual screening[J].Methods,2015,71:58-63. |
| [17] | CARHART R E, SMITH D H, VENKATARAGHAVAN R .Atom pairs as molecular features in structure-activity studies:definition and applications[J].Journal of Chemical Information and Computer Sciences,1985,25(2):64-73. |
| [18] | O’ BOYLE N M, BANCK M, JAMES C A,et al .Open Babel:an open chemical toolbox[J].Journal of Cheminformatics,2011,3:33/1-14. |
| [19] | GOBBI A, POPPINGER D .Genetic optimization of combinatorial libraries[J].Biotechnology and Bioengineering,1998,61(1):47-54. |
| [20] | SIEG J, FELDMANN C W, HEMMERICH J,et al .MolPipeline:a Python package for processing mole-cules with RDKit in scikit-learn[J].Journal of Chemical Information and Modeling,2024,64(24):9027-9033. |
| [21] | DUVENAUD D K, MACLAURIN D, IPARRAGUIRRE J,et al .Convolutional networks on graphs for learning molecular fingerprints[C]∥ Proceedings of the 29th International Conference on Neural Information Proce-ssing Systems.Cambridge:MIT Press,2015:2224-2232. |
| [22] | ZHAO W, WANG L, MIRJALILI S .Artificial hummingbird algorithm:a new bio-inspired optimizer with its engineering applications[J].Computer Methods in Applied Mechanics and Engineering,2022,388:114194/1-45. |
| [23] | DORIGO M, MANIEZZO V, COLORNI A .Ant system:optimization by a colony of cooperating agents[J].IEEE Transactions on Systems,Man,and Cybernetics,Part B (Cybernetics),1996,26(1):29-41. |
| [24] | WU J, CHEN Y, WU J,et al .Large-scale comparison of machine learning methods for profiling prediction of kinase inhibitors[J].Journal of Cheminformatics,2024,16:13/1-22. |
| [25] | JIANG D, WU Z, HSIEH C Y,et al .Could graph neural networks learn better molecular representation for drug discovery?A comparison study of descriptor-based and graph-based models[J].Journal of Cheminformatics,2021,13:12/1-23. |
| [26] | DE P, KAR S, AMBURE P,et al .Prediction reliability of QSAR models:an overview of various validation tools[J].Archives of Toxicology,2022,96:1279-1295. |
/
| 〈 |
|
〉 |