
收稿日期: 2025-01-17
网络出版日期: 2025-11-27
基金资助
国家自然科学基金项目(12322119);国家自然科学基金项目(12401630)
Prediction of Target Inhibitor Activity by Integrating Machine Learning and Metaheuristic Algorithms
Received date: 2025-01-17
Online published: 2025-11-27
Supported by
the National Natural Science Foundation of China(12322119)
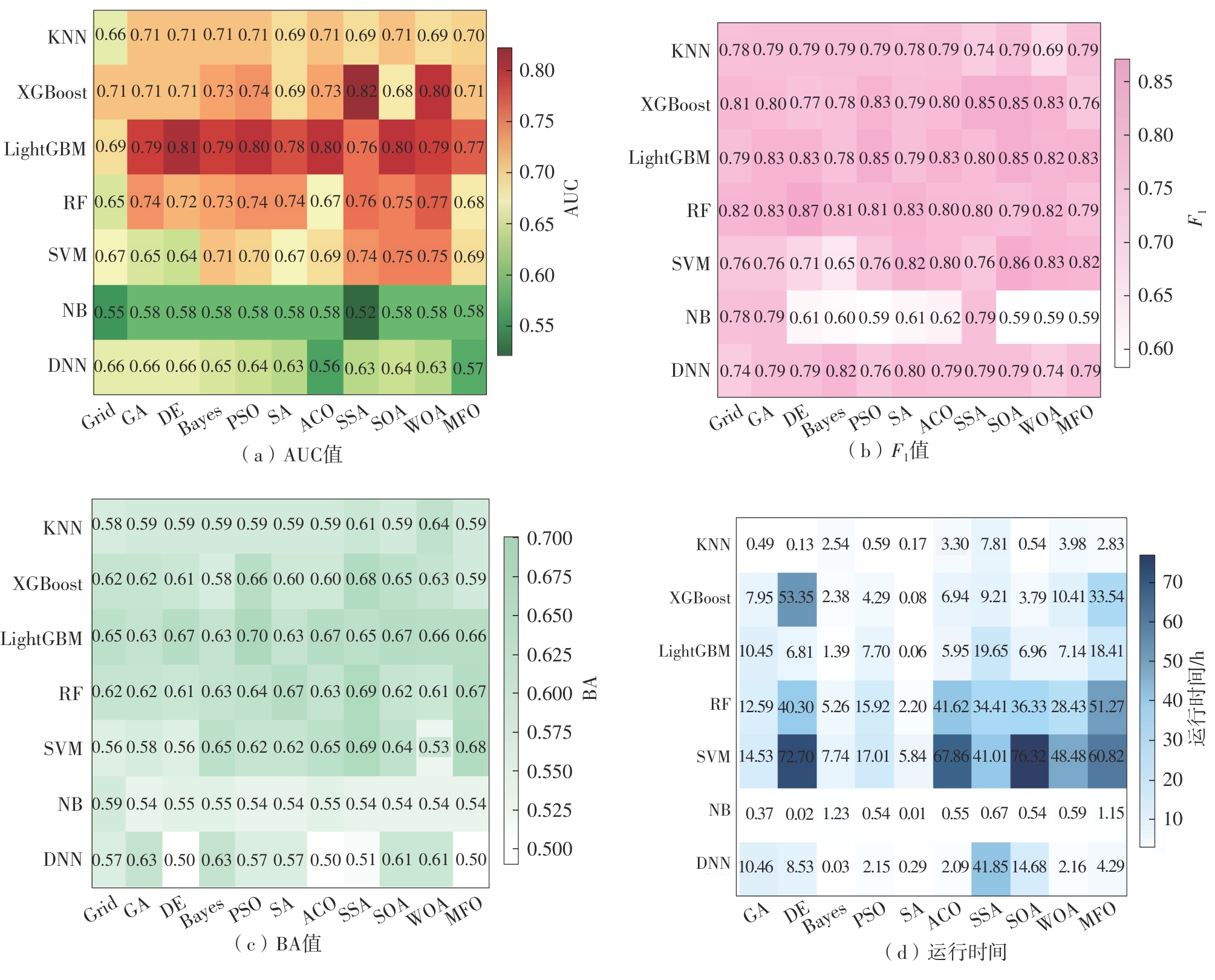
传统的机器学习(ML)和深度学习(DL)在预测靶点抑制剂的选择性方面发挥着关键作用。许多基于现有数据集的模型可用于预测化合物的生物活性,但对于ML和DL用于此类活性预测任务的性能孰优孰劣仍存在争议。该文基于不同分子特征构建数据集,运用10种元启发式算法优化11种ML和DL模型的超参数,旨在系统比较模型的预测性能,识别最优模型。结果表明,基于元启发式超参数优化算法的ML和DL模型,在预测性能上显著优于采用传统网格搜索优化超参数的ML和DL模型。此外,在低维特征空间中,基于分子图的DL模型(如SSA-GAT和SSA-Attentive FP)能够通过端到端的学习机制,自动从数据中提取有效特征,其性能优于ML模型;而在高维特征空间(如RDKit计算的ECFP、AtomPairs、MACCS指纹组合形成的特征空间)中,ML方法借助信息互补的分子特征和元启发式优化算法的高阶寻优能力,能够有效捕捉特征之间的复杂交互关系,通常在高维建模中展现出更高的准确性与鲁棒性。这些发现为指导选择ML和DL方法用于靶点抑制剂的活性预测提供了有用的信息。
凌飞 , 顾学荣 . 集成机器学习和元启发式算法的靶点抑制剂活性预测[J]. 华南理工大学学报(自然科学版), 2026 , 54(2) : 91 -101 . DOI: 10.12141/j.issn.1000-565X.250020
Traditional machine learning (ML) and deep learning (DL) play a key role in predicting the activity of target inhibitors. Many models based on existing datasets can predict compound bioactivity. However, debate persists regarding whether ML or DL performs better for such prediction tasks. In this study, datasets were constructed based on different molecular representations. Ten metaheuristic algorithms were applied to optimize the hyperparameters of eleven ML and DL models, aiming to systematically compare their predictive performance and identify the optimal ones. The results show that ML and DL models whose hyperparameters were optimized by metaheuristic algorithms significantly outperformed those optimized using the traditional grid search method. Furthermore, in low-dimensional feature spaces, graph-based DL models, such as SSA-GAT and SSA-Attentive FP, can automatically extract informative features from data via an end-to-end learning mechanism, yielding better performance than ML models. In contrast, in high-dimensional feature spaces (e.g., the feature space formed by combining RDKit descriptors with ECFP, AtomPairs, and MACCS fingerprints), ML methods, leveraging the complementary information in molecular features and the powerful optimization capability of metaheuristic algorithms, can effectively capture complex feature interactions. Consequently, ML methods often demonstrate higher accuracy and robustness in high-dimensional modeling. These findings provide valuable guidance for selecting between ML and DL approaches for target inhibitor activity prediction.
| [1] | FENG B, LIU Z, HUANG N,et al .A bioactivity foundation model using pairwise meta-learning[J].Nature Machine Intelligence,2024,6(8):962-974. |
| [2] | ZHANG H, MAO J, QI H Z,et al .Developing novel computational prediction models for assessing chemical-induced neurotoxicity using na?ve Bayes classifier technique[J].Food and Chemical Toxicology,2020,143:111513/1-11 |
| [3] | PRABHA N K, SHARMA A, SANDHU H,et al .TNFipred:a classification model to predict TNF-α inhibitors[J].Molecular Diversity,2024,28(3):1697-1707. |
| [4] | LI B, KANG X, ZHAO D,et al .Machine learning models combined with virtual screening and molecular docking to predict human topoisomerase I inhibitors[J].Molecules,2019,24(11):2107/1-16. |
| [5] | SHI J, ZHAO G, WEI Y .Computational QSAR model combined molecular descriptors and fingerprints to predict HDAC1 inhibitors[J].Medecine Sciences,2018,34:52-58. |
| [6] | KANG M G, KANG N S .Predictive model for drug-induced liver injury using deep neural networks based on substructure space[J].Molecules,2021,26(24):7548/1-16 |
| [7] | DENG J, YANG Z, WANG H,et al .A systematic study of key elements underlying molecular property prediction[J].Nature Communications,2023,14(1):6395/1-20. |
| [8] | LV Q, CHEN G, YANG Z,et al .Meta learning with graph attention networks for low-data drug discovery[J].IEEE Transactions on Neural Networks and Learning Systems,2023,35(8):11218-11230. |
| [9] | YANG K, SWANSON K, JIN W,et al .Analyzing learned molecular representations for property prediction[J].Journal of Chemical Information and Modeling,2019,59(8):3370-3388. |
| [10] | MASTROPIETRO A, PASCULLI G, BAJORETH J .Learning characteristics of graph neural networks predicting protein-ligand affinities[J].Nature Machine Intelligence,2023,5(12):1427-1436. |
| [11] | MAYR A, KLAMBAUER G, UNTERTHINER T,et al .Large-scale comparison of machine learning methods for drug target prediction on ChEMBL[J].Chemical Science,2018,9:5441-5451. |
| [12] | WAINER J, FONSECA P .How to tune the RBF SVM hyperparameters?An empirical evaluation of 18 search algorithms[J].Artificial Intelligence Review,2021,54(6):4771-4797. |
| [13] | BERGSTRA J, BENGIO Y .Random search for hyper-parameter optimization[J].Journal of Machine Lear-ning Research,2012,13(2):281-305. |
| [14] | WANG X, JIN Y, SCHMITT S,et al .Recent advances in Bayesian optimization[J].ACM Computing Surveys,2023,55(13s):1-36. |
| [15] | YAMASHITA R, NISHIO M, DO R K G,et al .Convolutional neural networks:an overview and application in radiology[J].Insights into Imaging,2018,9:611-629. |
| [16] | CERETO-MASSAGUé A, OJEDA M J, VALLS C,et al .Molecular fingerprint similarity search in virtual screening[J].Methods,2015,71:58-63. |
| [17] | CARHART R E, SMITH D H, VENKATARAGHAVAN R .Atom pairs as molecular features in structure-activity studies:definition and applications[J].Journal of Chemical Information and Computer Sciences,1985,25(2):64-73. |
| [18] | O’ BOYLE N M, BANCK M, JAMES C A,et al .Open Babel:an open chemical toolbox[J].Journal of Cheminformatics,2011,3:33/1-14. |
| [19] | GOBBI A, POPPINGER D .Genetic optimization of combinatorial libraries[J].Biotechnology and Bioengineering,1998,61(1):47-54. |
| [20] | SIEG J, FELDMANN C W, HEMMERICH J,et al .MolPipeline:a Python package for processing mole-cules with RDKit in scikit-learn[J].Journal of Chemical Information and Modeling,2024,64(24):9027-9033. |
| [21] | DUVENAUD D K, MACLAURIN D, IPARRAGUIRRE J,et al .Convolutional networks on graphs for learning molecular fingerprints[C]∥ Proceedings of the 29th International Conference on Neural Information Proce-ssing Systems.Cambridge:MIT Press,2015:2224-2232. |
| [22] | ZHAO W, WANG L, MIRJALILI S .Artificial hummingbird algorithm:a new bio-inspired optimizer with its engineering applications[J].Computer Methods in Applied Mechanics and Engineering,2022,388:114194/1-45. |
| [23] | DORIGO M, MANIEZZO V, COLORNI A .Ant system:optimization by a colony of cooperating agents[J].IEEE Transactions on Systems,Man,and Cybernetics,Part B (Cybernetics),1996,26(1):29-41. |
| [24] | WU J, CHEN Y, WU J,et al .Large-scale comparison of machine learning methods for profiling prediction of kinase inhibitors[J].Journal of Cheminformatics,2024,16:13/1-22. |
| [25] | JIANG D, WU Z, HSIEH C Y,et al .Could graph neural networks learn better molecular representation for drug discovery?A comparison study of descriptor-based and graph-based models[J].Journal of Cheminformatics,2021,13:12/1-23. |
| [26] | DE P, KAR S, AMBURE P,et al .Prediction reliability of QSAR models:an overview of various validation tools[J].Archives of Toxicology,2022,96:1279-1295. |
/
| 〈 |
|
〉 |
地址:广州 五山 华南理工大学17号楼 邮政编码:510640
电话: 020-87111794 邮箱:journal@scut.edu.cn

